
Potential Longevity Therapeutics: TEAD inhibitors and their role in cancer, fibrosis, and aging
Why a safe, balanced pan-TEAD inhibitor may be a pipeline in a product spanning oncology, fibrosis, and aging

For over two decades in this field, I have watched the pharmaceutical industry treat certain targets the way a sailor treats a reef: charted, feared, and given a wide berth. TEAD is one of those reefs. It sits at the bottom of one of the most consequential signaling pathways in human biology, it is implicated in cancers that kill quickly and fibroses that kill slowly, and it has wrecked program after program of well-funded, well-staffed drug discovery teams. And yet I am more convinced than I have ever been that TEAD is not a reef to be avoided. It is a channel to be navigated, and the ships that learn to navigate it will reach places that matter enormously for both oncology and human aging.
Let me be direct about why I am writing this now. The data have shifted. We have a clinical frontrunner showing durable disease control with a manageable safety profile, and we have a graveyard of more aggressive approaches that failed in ways that, in retrospect, were predictable from the biology. The question with TEAD is no longer whether modulating it does something therapeutically meaningful; it does. The question is whether we can do it safely. That distinction, between efficacy and tolerable efficacy, is the entire game here, and it is the lens through which the rest of this piece is written.
Why TEAD Sits at a High-Value Node of Biology
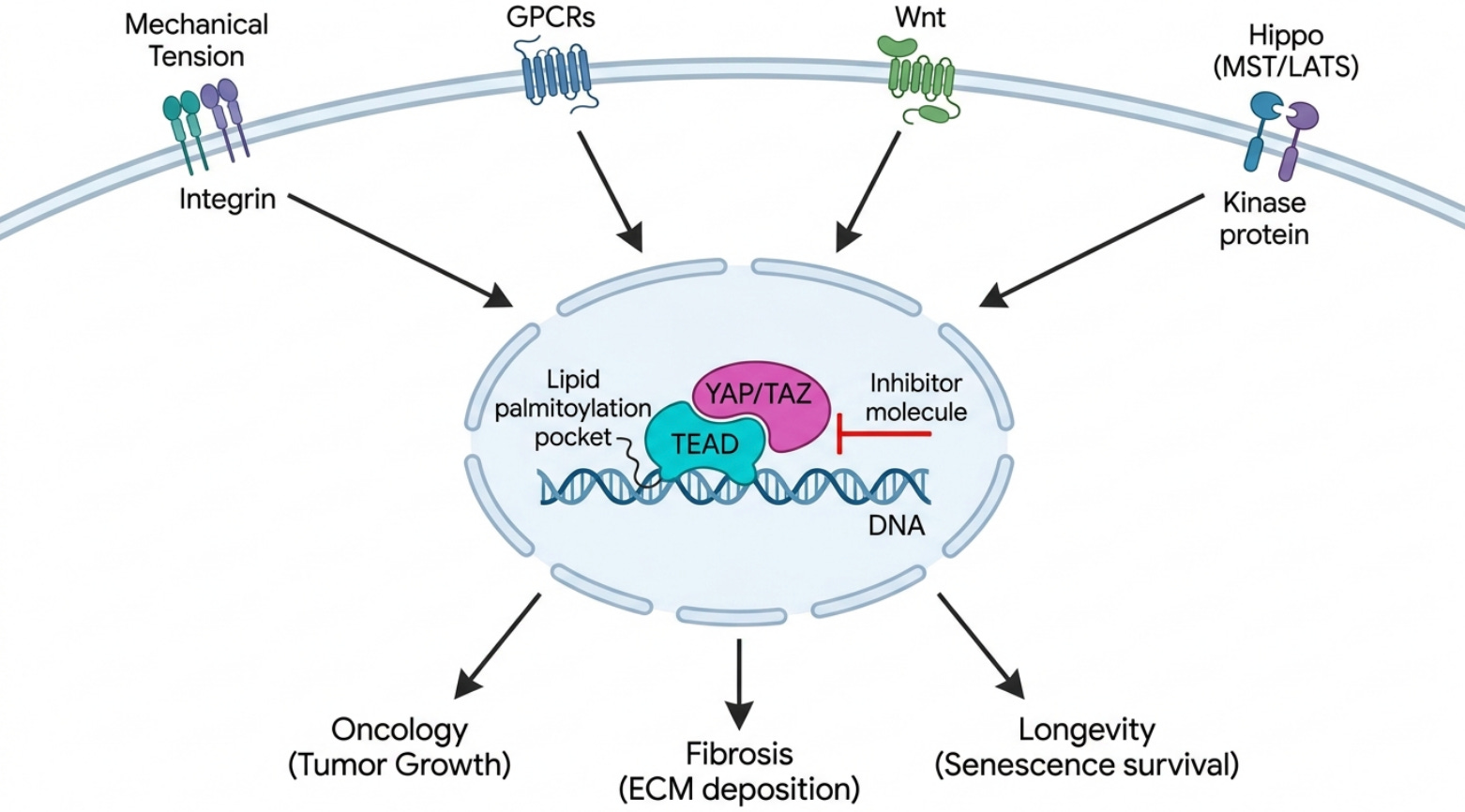
To understand why so many companies keep returning to TEAD despite the casualties, you have to understand where it lives. TEAD1, TEAD2, TEAD3, and TEAD4 are the terminal DNA-binding transcription factors of the Hippo pathway. On their own they are largely inert; they cannot drive transcription without their coactivators YAP and TAZ. The Hippo kinase cassette, principally MST1/2 and LATS1/2, normally restrains YAP and TAZ by phosphorylating them and keeping them out of the nucleus. When that restraint is lifted, whether through loss of the tumor suppressor NF2 (Merlin), inactivating mutations in LATS, or mechanical cues from a stiff extracellular matrix, YAP and TAZ translocate into the nucleus, dock onto TEAD, and switch on a program of growth, regeneration, and stemness. The canonical readouts of this program, CTGF/CCN2 and CYR61/CCN1, are among the most reliable transcriptional fingerprints in all of cell biology.
What makes TEAD genuinely druggable, and not merely interesting, is a quirk of its structure. Each TEAD paralog carries a central lipid pocket and undergoes auto-palmitoylation on a conserved cysteine, covalently attaching a palmitate to itself. That pocket is the main pharmacological handle on the protein. The alternative site, the YAP-to-TEAD protein-protein interface, is large, flat, and shallow, with the critical contact at what the field calls Interface 3, and it was historically considered undruggable for exactly those reasons. So the lipid pocket is where most of the action is, and it is a defined, lipophilic, enclosed space; the kind of site that structure-based and AI-driven design handles well.
The complication is paralogy. There are four TEAD proteins with overlapping but tissue-biased expression and a high degree of functional redundancy. If you block one paralog cleanly, the others often compensate, and your beautiful selective inhibitor produces disappointing biology. This single fact, the redundancy of four paralogs, explains a great deal of what has gone wrong clinically, and it points directly at what should go right.
Why TEAD Kept Failing
The first crude attempt at this target was verteporfin, a photosensitizer repurposed to disrupt YAP/TEAD. It was a blunt instrument with poor selectivity and properties that made it unsuitable as a systemic oncology agent, but it proved the concept that interfering with this axis does something. The serious medicinal chemistry that followed ran into two recurring walls, and both are mechanistic rather than accidental.
The first wall is renal toxicity. Proteinuria and albuminuria show up across TEAD inhibitor programs to the point where I regard them as a class effect rather than a compound-specific liability. The mechanism is not mysterious: TEAD1 and YAP are functionally important in kidney podocytes and in maintaining glomerular integrity. Suppress that biology hard enough and the filtration barrier leaks. The second wall is cardiac. TEAD1 is essential in cardiomyocytes, and deep blockade raises real concerns about cardiotoxicity and QTc prolongation. These are not safety signals you can engineer away with a better salt form. They are the consequence of inhibiting a target that healthy adult tissues genuinely need.
The clinical record makes the point with uncomfortable clarity. Novartis advanced IAG933, a direct YAP/TAZ-TEAD Interface-3 protein-protein interaction disruptor with pan-TEAD activity, into a Phase 1 study in NF2- and LATS-altered mesothelioma. The data presented at ESMO 2025 showed a modest objective response rate in the range of thirteen to roughly seventeen percent in pleural mesothelioma, and the dose-limiting toxicities were precisely the two predicted by the biology: QTc prolongation on the cardiac side and Grade 2 proteinuria on the renal side. The program was discontinued late in 2025 for limited activity combined with a narrow therapeutic window. Ikena Oncology took the opposite chemical strategy with IK-930, a TEAD1-selective inhibitor, and discontinued it in May 2024 after disappointing Phase 1 efficacy, then exited oncology altogether. SpringWorks halted SW-682. Three distinct strategies, three failures, and a pattern worth reading carefully.
The Case for a Gentle, Balanced Pan-TEAD Inhibitor
Here is where the biology and the clinical history converge into a thesis. Deep single-paralog TEAD1 blockade is the worst of both worlds: it over-hits the precise heart and kidney tissues where TEAD1 is most essential, and it simultaneously leaves TEAD2, TEAD3, and TEAD4 free to compensate in the tumor. So you accumulate the toxicity of hitting TEAD1 without capturing the full antitumor effect of shutting the program down. Deep, complete pan-TEAD shutdown, the IAG933 approach, captures the program but collapses the therapeutic window, because now you are maximally ablating TEAD1 in cardiomyocytes and podocytes at the same time as you hit the tumor. Both failure modes are coherent. Neither is bad luck.
The profile that should work is partial, balanced suppression across all four paralogs. You want enough inhibition to flip the oncogenic and profibrotic transcriptional state, but not so much that you ablate the physiological TEAD1 that the heart and kidney depend on. Evolution is a cruel master here; it built TEAD1 into both pathology and essential homeostasis, and it offers no clean separation between the two. So the separation has to come from pharmacology. The levers are concrete. You tune systemic exposure for partial modulation rather than maximal target engagement. You favor reversible, non-covalent pocket binders, because reversibility allows titration in a way covalent inactivation does not. You consider intermittent dosing schedules that give the essential tissues windows of recovery. And you deliberately balance the paralog affinities so that TEAD1 is not the paralog you hit hardest. This is a design problem with a defined binding pocket, which is exactly the kind of problem modern generative and structure-based methods are built to solve.
TEAD in Cancer
The cleanest case for TEAD in oncology is genetic dependency. When NF2/Merlin is lost or LATS1/2 is mutated, YAP/TAZ-TEAD signaling becomes constitutively active, and the tumor becomes addicted to that signal. Mesothelioma is the lead indication precisely because roughly forty to fifty percent of cases carry NF2 alterations, which gives you a biomarker-defined population enriched for response. Beyond mesothelioma, epithelioid hemangioendothelioma is driven by a TAZ-CAMTA1 fusion that funnels straight through this axis, NF2-associated meningioma and schwannoma share the dependency, and a subset of NSCLC carries the relevant lesions.
There is a second, arguably larger, opportunity in resistance. TEAD reactivation is a documented mechanism of acquired resistance to EGFR inhibitors, to KRAS(G12C) inhibitors, and to BRAF/MEK (MAPK) pathway inhibitors. Tumors under pressure from targeted therapy frequently escape by turning the YAP/TAZ-TEAD program back on. That creates a combination rationale that extends TEAD far beyond the NF2-mutant niche, into common solid tumors where the standard of care eventually fails through this exact escape hatch. A well-tolerated pan-TEAD agent that can be dosed alongside a MAPK inhibitor without compounding toxicity is a different commercial and clinical proposition than a single-agent rare-tumor drug.
TEAD in Fibrosis
If oncology is where TEAD earned its reputation, fibrosis is where I think it earns its breadth. YAP/TAZ-TEAD is the master mechanotransduction switch for myofibroblast activation. The logic is a feed-forward loop that is almost diabolical in its efficiency: injury or stiffness in the extracellular matrix is sensed mechanically, YAP and TAZ go nuclear, TEAD switches on the profibrotic program (CTGF/CCN2 is a direct TEAD target), the cell lays down collagen and turns on alpha-SMA, the matrix gets stiffer, and the stiffer matrix drives still more nuclear YAP/TAZ. Once that loop is running, it tends to run on its own.
This biology is not specific to one organ, which is the point. The same switch underlies idiopathic pulmonary fibrosis, liver fibrosis and MASH, kidney fibrosis in chronic kidney disease, cardiac fibrosis, and the fibrotic burden of systemic sclerosis. My company has already shown that an AI-discovered target paired with an AI-designed molecule can reach the clinic in fibrosis, with our generative-AI TNIK inhibitor reaching Phase 2a in IPF, published in Nature Medicine. That experience taught me something relevant to TEAD: in fibrosis, the molecules that matter are the ones that interrupt the upstream master switches of myofibroblast identity, not the ones that mop up a single downstream mediator. TEAD is about as upstream and as master a switch as the fibrosis field has.
Possible Role of TEAD in Aging and Longevity
Now extend the logic across a lifetime. Tissues stiffen with age. Extracellular matrix accumulates and crosslinks, collagen content and rigidity rise, and that mechanical change is not cosmetic; it is a chronic, ambient signal that keeps stromal YAP/TAZ-TEAD partially activated across many organs. What we call fibrosis in a disease label is, in aged tissue, a slow, diffuse, organ-spanning process I think of as the fibrosis-of-aging. It contributes to the stiffening of vasculature, the loss of organ compliance, and the gradual replacement of functional parenchyma with scar.
There are also context-dependent connections between this axis and cellular senescence, and here the mechanism is more concrete than most people appreciate. Senescent cells are protein-secretion factories, churning out the inflammatory SASP payload, and that places an enormous folding burden on the endoplasmic reticulum. Recent whole-genome CRISPR work showed that the YAP-TEAD complex keeps these cells alive by repressing DDIT4, an endogenous mTOR inhibitor; with DDIT4 held down, mTOR stays active, the ER keeps expanding, and the cell can process its SASP load without tipping into lethal ER stress. Block TEAD and that repression lifts: DDIT4 rises, mTOR falls, ER biogenesis stalls, and the senescent cell, still committed to synthesizing its payload but now without the capacity to fold it, dies of proteotoxic stress. Healthy cells, which do not carry that secretory burden, are largely spared. In aged mice, TEAD inhibition with verteporfin cleared senescent cells, reduced age-related lung fibrosis, and restored tissue homeostasis. That is a genuinely selective, mechanism-based route to senolysis, and it sits right next to the fibrosis and stiffening biology rather than being a separate story.
None of this is a recent hunch, and it is worth grounding in the prior literature because the case has been building quietly for more than a decade. The Hippo pathway, whose sole nuclear output is the YAP/TAZ-TEAD transcriptional complex, was flagged as dysregulated in both aging and cancer in a widely cited 2019 review, and its dual involvement in tissue regeneration and organismal aging was mapped the following year. The single most important paper for anyone building a TEAD inhibitor with longevity intent showed, in a 2023 Nature Aging study, that the YAP-TEAD complex actively promotes the survival of senescent cells by lowering endoplasmic reticulum stress, the mechanistic result underneath the DDIT4 and mTOR story above. The mechanotransduction link makes the tie to aging tighter still: aged extracellular matrix stiffens, and stiffness is precisely the mechanical signal that drives YAP into the nucleus to partner with TEAD, a feed-forward loop that converts stem cells to a fibrogenic fate in aged tissue. The picture is genuinely context-dependent and I want to represent it honestly, because in some compartments YAP activity is protective, alleviating senescence and osteoarthritic degeneration and acting as a geroprotective factor in primate gingival aging. This is exactly why an ablative, oncology-grade TEAD inhibitor is the wrong tool for aging and a gentle, balanced, partial pan-TEAD modulator is the right one: the goal in longevity is not to shut the pathway off, which would cripple the regenerative arm the tissue still needs, but to lower the chronic pathological tone that accumulates with stiffening matrix and senescent-cell persistence while leaving physiological turnover intact.
I will not overstate these links; they are more contingent than the hard genetic dependencies in cancer, and the human data are not yet in for this mechanism. But the directional case is strong, and it has an important design implication. For aging, you absolutely do not want an ablator. You want a balanced modulator that lowers the chronic profibrotic tone and can tip SASP-dependent senescent cells over the edge, without shutting down the YAP/TAZ-TEAD signaling that healthy tissues still need for normal maintenance and repair, and while preserving the regenerative YAP/TAZ activity that actually declines in some aged stromal compartments. The safety thesis I described for oncology is not a constraint in the longevity setting; it is the entire requirement.
A Pipeline in a Product
I have written before about the idea of a single target that behaves like a Swiss Army knife, one mechanism that opens onto many indications across the disease-to-aging continuum, much as I have framed NLRP3 as a multi-indication longevity target. A safe, balanced pan-TEAD inhibitor fits that pipeline-in-a-product framing more naturally than almost any target I can name, because the same molecular event, partial suppression of the YAP/TAZ-TEAD transcriptional program, is therapeutic in three distinct domains.
The indications sort into a logical sequence by risk and timeline:
Near-term oncology. NF2/LATS-altered mesothelioma as monotherapy (biomarker-defined dependency); epithelioid hemangioendothelioma (TAZ-CAMTA1 fusion funnels through TEAD); NF2 meningioma and schwannoma (shared Merlin-loss dependency); EGFR-, KRAS-, and MAPK-resistant solid tumors in combination (TEAD reactivation as a resistance mechanism).
Mid-term fibrosis. IPF (myofibroblast mechanotransduction switch); liver fibrosis and MASH (stellate-cell activation); CKD and renal fibrosis (here the kidney safety thesis matters most); cardiac fibrosis (with the same caveat for the heart); systemic sclerosis (multi-organ profibrotic program).
Longer-term age-related. Fibrosis-of-aging across organs (chronic stromal YAP/TAZ-TEAD tone); tissue and vascular stiffening (the mechanical feed-forward loop); senescence- and SASP-associated pathology as an adjunct to senotherapeutics.
The reason a single safe pan-TEAD agent can credibly span all three of these domains is that the underlying biology does not change as you move from a mesothelioma to a fibrotic liver to an aged blood vessel; what changes is only the source of the YAP/TAZ-TEAD activation and the degree of suppression you want. In cancer the source is genetic and the desired suppression is deep but window-limited; in fibrosis the source is mechanical and the desired suppression is sustained but moderate; in aging the source is the ambient stiffness of an old matrix and the desired suppression is gentle and chronic. One palmitate-pocket-binding molecule, designed for balanced partial pan-TEAD inhibition with reversible kinetics and a dosing schedule that protects heart and kidney, is in principle deployable across that entire spectrum simply by changing the dose, the schedule, and the patient population. That is what I mean when I say pipeline in a product, and it is the same Swiss Army knife logic that makes a small number of well-chosen targets worth far more, scientifically and economically, than the indication-by-indication grind the industry usually accepts.
The Graveyard: What the Failures Taught Us
Before I map the live field, it is worth stopping at the graveyard, because in TEAD the failures are more instructive than most successes. Every one of these programs died for a mechanistic reason that was legible in the biology beforehand, and together they define the design constraints the winning molecule has to respect.
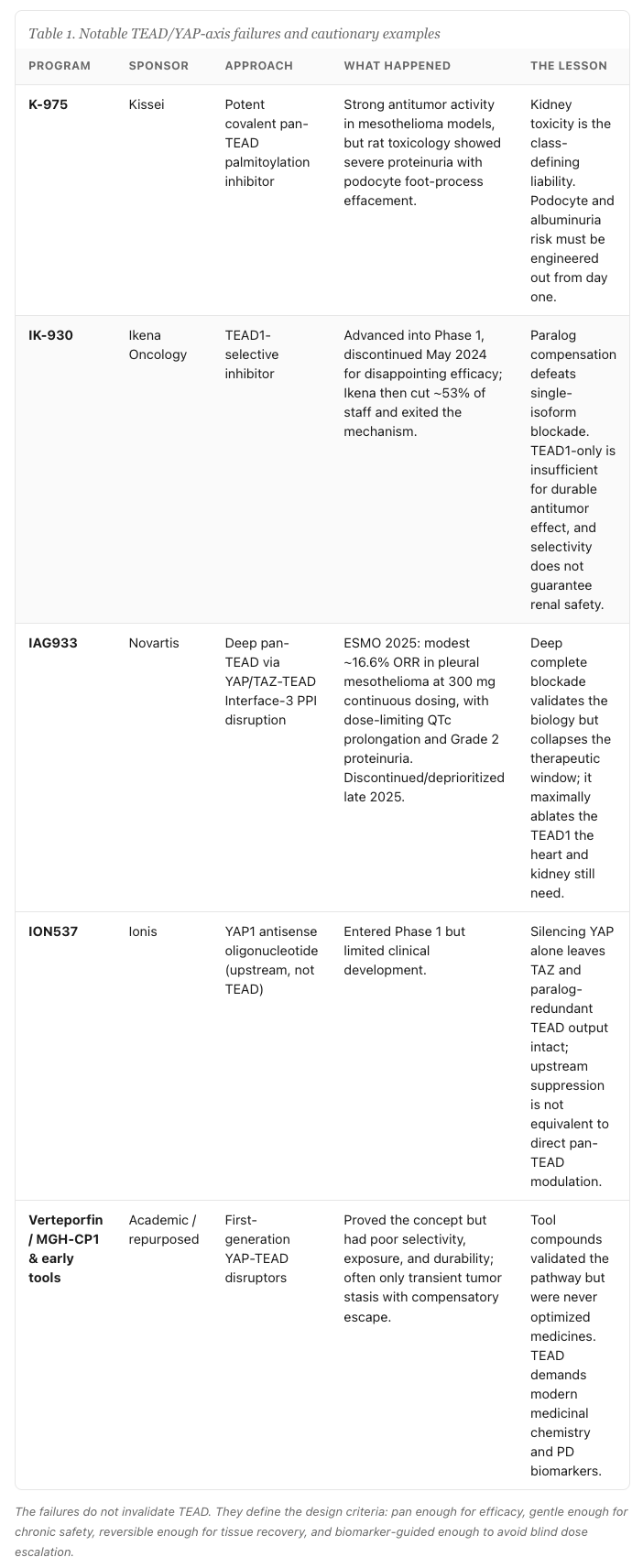
The pattern is impossible to miss. Deep single-paralog selectivity fails on efficacy. Deep complete pan blockade fails on the therapeutic window. Upstream YAP-only silencing fails on redundancy. Three distinct strategies, three distinct failure modes, and every one of them coherent from the biology rather than the product of bad luck.
The Living Field: Current Competitive Landscape
Now line up the programs that are still standing, and the story resolves into a single clear signal.
The frontrunner is Vivace Therapeutics’ VT3989, a first-in-class TEAD auto-palmitoylation inhibitor that binds the lipid pocket and acts as a pan-TEAD agent. In its Phase 1/2 study in advanced solid tumors, including NF2-mutant refractory mesothelioma, the data reported in Nature Medicine in 2025 and updated at ESMO showed, in the optimized-dose mesothelioma cohort (22 patients, 2-weeks-on / 2-weeks-off dosing with UACR-guided adjustments), a 32% objective response rate, an 86% disease control rate, and a median progression-free survival of roughly forty weeks, with safety driven mainly by manageable low-grade proteinuria and fatigue. It carries FDA Orphan Drug and Fast Track designations and is advancing toward a registrational Phase 3 in mesothelioma. I want to be precise about why this matters: VT3989 is partial, it is pan, and its main toxicity is the expected low-grade proteinuria rather than a window-collapsing combination of QTc and renal injury. Its profile is the clinical validation of the gentle, balanced pan-TEAD thesis.
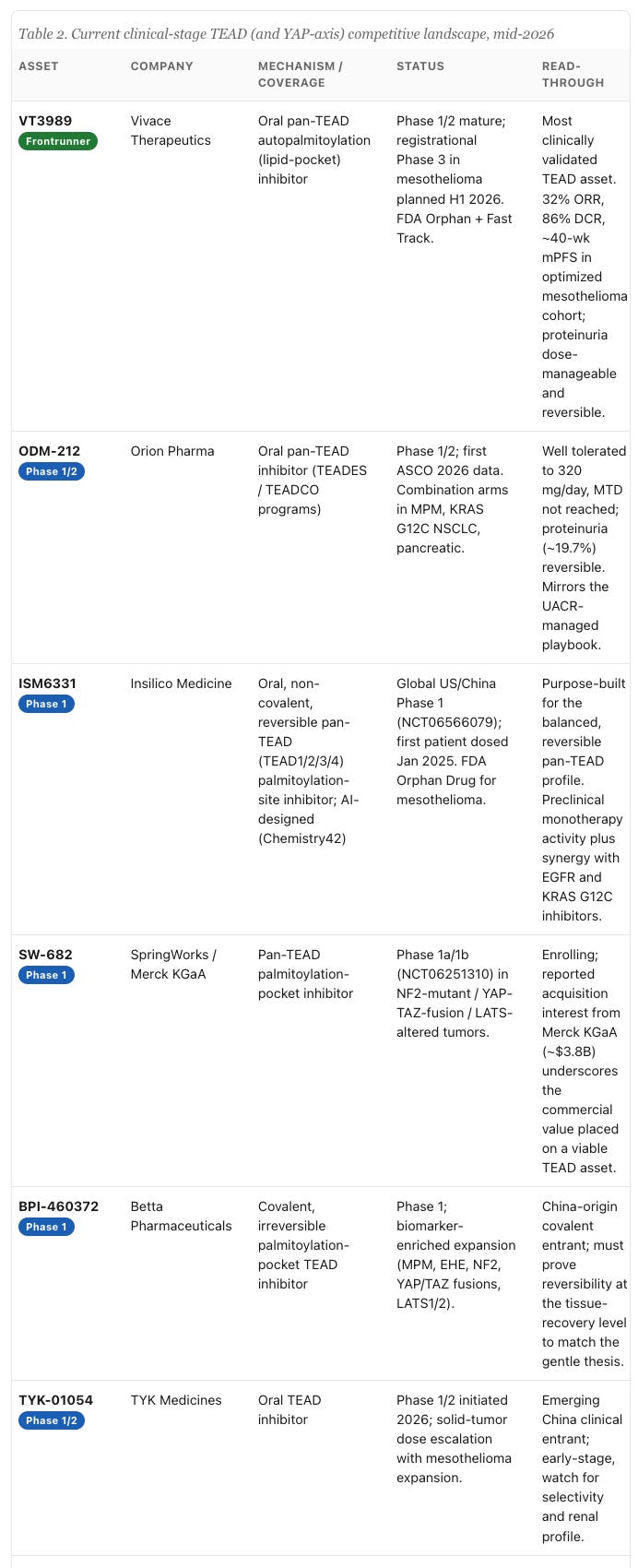

Beyond the clinic, the preclinical and IND-stage field is broader still, and it is worth noting because it shows where the next wave is heading. Genentech’s GNE-7883 is an allosteric pan-TEAD tool compound that has provided some of the cleanest evidence that pan inhibition overcomes KRAS(G12C)-inhibitor resistance, reinforcing the combination rationale. Signet Therapeutics and XtalPi’s SIGX2649, an AI/structure-enabled pan-TEAD inhibitor that also recruits the VGLL4 co-repressor, filed its IND in early 2026 with a renal-safety differentiation claim. Sporos BioDiscovery is pursuing broader TEAD1/4 coverage, Betta and Zai Lab and others populate a rapidly globalizing, increasingly China-heavy IP landscape, and Boehringer Ingelheim and the Walter and Eliza Hall Institute are bypassing enzymatic inhibition entirely with bifunctional PROTAC degraders that recruit IAPs to physically destroy the TEAD protein. The influx of AI-designed and structure-enabled programs, and the recurring emphasis on renal safety as the differentiator, both tell you the field now agrees on what the hard part actually is.
The strategic read is hard to miss when you line the living field against the graveyard. The one program with durable activity and a livable safety profile, VT3989, is a partial, balanced, lipid-pocket pan-TEAD agent, and the programs advancing behind it, ODM-212, ISM6331, SW-682, cluster around the same reversible, tunable, UACR-guided profile. The field is converging, through expensive trial and error, on the conclusion that balanced, safety-optimized pan-TEAD inhibition beats both extremes. That convergence is the opportunity, because it tells you exactly what to design toward.
The Future of TEAD
The defined palmitate pocket is, for once, a target that plays to the strengths of computational drug design. It is enclosed, lipophilic, and structurally well characterized across all four paralogs, which means the central challenge, engineering a molecule that achieves balanced partial affinity across TEAD1 through TEAD4 with reversible kinetics and properties that permit intermittent dosing, is fundamentally a multi-parameter optimization problem. That is the regime where AI-driven generative chemistry earns its keep, designing not for maximal potency against one paralog but for a carefully shaped affinity profile that respects the heart and kidney. And it should be paired with pharmacodynamic biomarkers rather than blind dose escalation: the YAP/TAZ-TEAD output genes, CTGF/CCN2, CYR61/CCN1, ANKRD1, AXL, and LOX, give you a transcriptional readout of how much of the program you have actually silenced, while UACR and albuminuria give you the renal early-warning system. A TEAD program that escalates dose until proteinuria appears, instead of titrating to a biomarker of target engagement, is repeating the mistake of treating a homeostatic transcription factor like a mutant kinase. I have argued elsewhere, in Toward Pharmaceutical Superintelligence, that the real value of these methods is in solving exactly this kind of constrained, safety-bounded design problem rather than chasing raw potency, and TEAD is close to an ideal test case.
I want to be careful about what I am claiming. TEAD has humbled smarter and richer teams than most, and the longevity rationale in particular rests on links to senescence and stiffening that are real in direction but not yet proven in the clinic for this mechanism. What I am confident about is narrower and better supported: the genetic dependency in NF2/LATS cancers is validated, the fibrosis biology is among the most upstream switches available, and a frontrunner with a manageable safety profile has shown that the window the failed programs lacked actually exists if you aim for partial, balanced inhibition. Those three facts together justify treating a safety-optimized pan-TEAD inhibitor as one of the more compelling pipeline-in-a-product opportunities spanning oncology, fibrosis, and aging.
A standing disclaimer, because it matters: nothing here is investment advice, and the specific companies, programs, and drugs I have named are discussed only for scientific and strategic analysis, not as recommendations of any kind. My interest in TEAD is in the biology and the chemistry, and in the broader point that the targets worth pursuing are the ones where the hard problem is not whether the mechanism works, but whether we can engineer it to work safely enough to use across a human lifespan. TEAD is squarely one of those targets, and I expect the next few years of data to make that case far more concrete than I can today.
Disclaimer: This article is written with the help of generative tools so beware of hallucinations. The images were generated using NanoBanana. Don’t buy, sell any securities, or take any drugs based on this article or any of its contents. The information and views expressed in this article are for informational and educational purposes only and do not constitute medical advice. The content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or another qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this article. The author is sharing personal experiences and opinions. These experiences are not a recommendation or endorsement for any specific treatment, drug, or course of action. The medications and therapeutic strategies discussed may not be suitable for everyone and can have significant risks and side effects. Some of the drugs mentioned are investigational and have not been approved by the FDA or other regulatory agencies for the uses discussed. While the author is the CEO of Insilico Medicine, the statements and view presented in Forver.ai do not represent the views and opinions of Insilico Medicine.