
Can We Accelerate Drug Discovery and Longevity Research by Replacing/Augmenting IND-Enabling Studies With Human Volunteers in Emerging Geographies?
Big Idea: Can UAE, KSA, India, and Other Countries Aspiring to Enter The Biotechnology Industry Leapfrog Established Players by Replacing GLP Animal Experiments with Expert Human Volunteers?

TL;DR: “For super-safe drugs, and above all for longevity therapeutics that must be safe by definition, we can treat the first human dose after non-GLP toxisity studies and give a year back to patients.”
Disclaimer: A disclaimer before you read further: these are my own hypothetical thoughts. They do not reflect the position, research, or work of my company or any of its partners. We are not working on replacing IND-enabling studies with human volunteers or with AI. I used AI to help with the research and some of the images, so if something here is wrong, blame the machine and comment below so I can fix it.
This article contains some insights and experience I picked up over the past five years working very hard to set new records in drug discovery and development productivity. Since we started our own drug discovery, in just past 5 years, we nominated 30 developmental candidates 12 of which reached clinical stage and multiple candidates and clinical assets were out-licensed to the pharmaceutical companies - meaning they met or exceeded their internal standards.
Our ideal business model is to discover and develop the drug using AI and automation to the level of Preclinical Candidate (PCC) for a specific target and license it out to the pharmaceutical company in need of this target. For this purpose, our typical PCC package contains 28-day non-GLP toxicity studies in at least two species (e.g. dog and monkey) and often three or more species if the target is competitive. When the target and/or the molecule is too novel, which is very often, the pharmaceutical companies prefer to wait and see how this or competing molecule performs in the clinic and we need to do the IND-enabling studies and even enter human clinical trials. IND-enabling studies are required to enter human clinical trials.
Like SpaceX developed the very reliable launch platform, we managed to industrialize the discovery process going from 0 to PCC as quick as 9 months with a moderately novel target. Think about this - about 4-5 months of this process is spent on discovery chemistry, in vitro and in vivo efficacy testing - the rest is 28-day non-GLP tox in several species often in parallel in order to nominate the PCC. Yes, it may sound fast - 28-day toxicity study may seem to last jus 1 moth, but in reality, with the preparations and with the data analysis, repetitions and cross-species comparison, it usually takes 4-5 months in non-GLP tox setting. And then you repeat this process in IND-enabling studies in GLP setting and it will cost you 3-10 times more than non-GLP tox and take you 9-12 months. Yes, it is not intuitive that 28-day process (sounds like 1 month) needs to take 12 months but it is the reality of life. Regardless of where you go, China, Europe, US, any GLP tox vendor will take approximately this long.
Here is the big secret and reveal - in drug discovery and development, AI helps accelerate the process from 0 until PCC and most of the acceleration comes in target discovery and generative chemistry. You can compress the in-vitro work with automation and massive parallelization in contract research organizations (CROs) but after PCC - it is super regulated territory. Before PCC - you “design the gun and the bullet” at PCC - you fire this bullet. You can navigate it with a better biomarker to target the right patients but AI can not make it go faster.
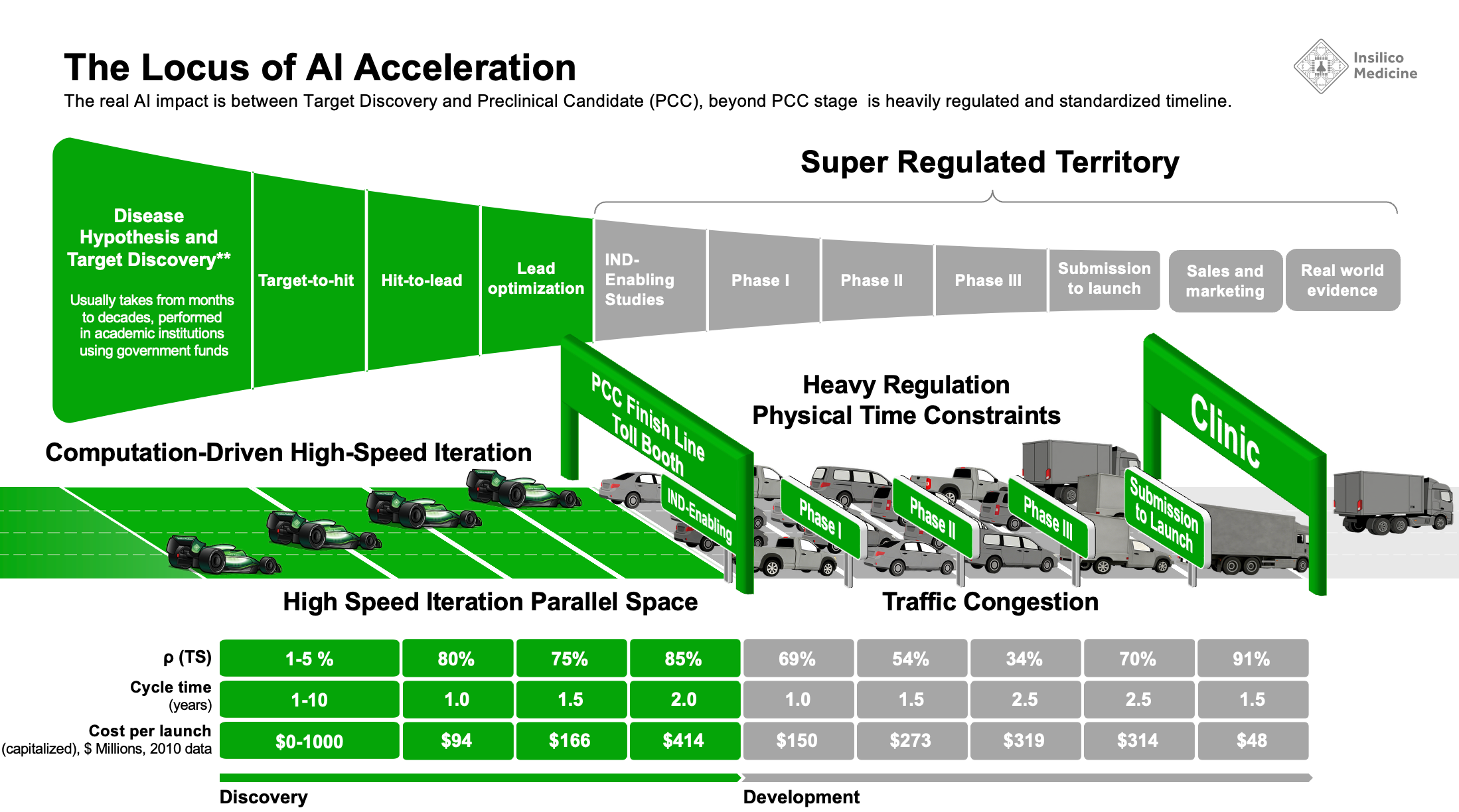
Another analogy are sports cars - you can compete with the others who can go from 0 to 100 faster but once you get to 100 on a highway, you will need to move with the speed of traffic and follow the signs set by the regulators. I think I coined this analogy. Anyone telling you that the AI startup can leapfrog big pharma in clinical trials with AI must show a few successes - so far I have not seen any acceleration on this front, quite the opposite.
The Hallway Meeting During FII Japan
One of my favorite conference series is the Future Investment Initiative (FII). It does not have the media circus of Davos. It is quieter and, to me, more serious, because it gets close to the biggest opportunities in human development, and some of my most important technology partnerships were forged in its hallways. FII in Riyadh became my annual pilgrimage. When schedule allows, I join the regional events too. You meet people there you would never meet at a scientific meeting.
At the recent FII in Japan, I spent time with leaders, investors, and media. The encounter I keep replaying happened in a hallway. I did not know, at first, that the man I was chatting with ran some of the most advanced hospitals in the MENA region. I will not name him; I did not ask his permission (yet). His Excellency told me his hospitals had excellent clinical trial facilities and that perhaps we could run our late-stage trials there.
I told him the truth, which is not flattering to the region’s reflex. No country in the Middle East is going to out-compete the United States, Australia, China, or even Europe on conventional clinical trial services. The market for most products is too small. And yet every MENA country I visit wants to become a contract research organization, preclinically or clinically, in a lane where there is almost no chance of offering a better, fully integrated service than what already exists.
Then I said the thing I had been circling for months. Nobody in the region has looked at the drug discovery process itself, found the one bottleneck nobody wants to touch, and decided to own it. There is a place in the pipeline where the most efficient companies on earth, including the Chinese, would gladly fly to save half a year. That place is not another CRO lane. It is the missing bridge between an AI-designed molecule and human biology.
That the conversation happened in Japan, the most aged society on earth, is something I will return to. The geography was not incidental.
The GLP Tox Tax, Counted in Months and Millions
If the science argues for early human data, the barrier is almost entirely regulatory and economic. I call it the GLP tox tax, a surcharge of time and capital that, for certain classes of drugs, buys almost no additional patient safety. Think about the next-generation GLP1 drug. The target is safe and the molecule to be competitive must be even safer than the previous generation and must have additional unique properties. And imagine that the drug you are trying to develop has been tested in at least 3 animal species alongside every possible competing molecule (usually, you need to demonstrate superior properties in apples-to-apples comparison) in 28-day non-GLP toxicity studies that include monkeys. Guess what - after this extensive testing that takes you four or five months or even longer, you still need to do another year of studies repeating what you did in the unregulated testing in a very regulated GLP manner in order to apply for human IND and to validate these new improved properties in real humans for a very safe target with a supposedly safer molecule. This step adds a year and about $2-4 million to the therapeutic program.
You have to separate two things people constantly conflate: the science of toxicology and the regulation called Good Laboratory Practice (GLP). They are not the same thing, and the gap between them is where a year goes to die.
Take a twenty-eight-day repeated-dose toxicology study. The name suggests one month. It is not one month. In a high-quality non-GLP setting, that single study runs roughly four to five months end to end, and every step is real work. You design the protocol and run dose-range-finding to pick the doses. You formulate the test article and analyze it to confirm concentration and stability. You run the in-life dosing phase, the twenty-eight days where animals actually receive the drug and are observed daily. Then comes necropsy, then histopathology read by board-certified pathologists, then toxicokinetics to understand exposure, then data analysis and cross-species comparison, then the repeats and confirmations that any honest program requires. We typically run this in two or three species in parallel, a rodent like the rat plus a non-rodent like the dog or the cynomolgus monkey, because regulators and biology both demand it. Four to five months, done well, done fast.
Now do the same study to full GLP standard. The science does not change. The species do not change. The dosing protocols, the clinical pathology, the pathologists are frequently the same. What changes is that the calendar stretches to nine to twelve months end to end and the cost rises by three to ten times. The extra time and money do not buy you more scientific signal. They buy a quality-assurance and documentation layer: independent QA audits of every single step, fully validated software with audit trails for every data point under 21 CFR Part 11, strict chain-of-custody, standardized and archived specimens, and final study reports that can run from many hundreds to over a thousand pages. Add vendor slotting and queue times, add QA review cycles, and the months pile up. This calendar is similar whether you run in China, in Europe, or in the United States. Nobody has found a shortcut, because the shortcut is not in the science. It is in the notarization.
The dollar figures, in round numbers and program-dependent, tell the same story. A non-GLP twenty-eight-day two-species package might run on the order of a few hundred thousand dollars. The full GLP IND-enabling package, meaning twenty-eight-day or longer GLP tox in two species plus the safety pharmacology core battery, genotoxicity, toxicokinetics, and the rest, commonly runs into the low single-digit millions and up. These are approximate. Your mileage varies by molecule, modality, and species. But the order of magnitude holds.
Here is the scientific point I keep coming back to. For a super-safe, well-precedented molecule, the non-GLP study already contains essentially all the decision-relevant safety information. The GLP repeat is a year-long, multimillion-dollar notarization of a conclusion you already hold. For a novel or high-risk molecule, that conservatism is appropriate and should stay exactly where it is. I am not arguing to dismantle a system that protects people. I am arguing that we have been applying maximum scrutiny uniformly, including to the cases that least need it, and patients are paying the interest on that tax in years of their lives.
This is where I lose patience. Not because regulators are stupid. They are not, and I respect the FDA’s caution. It is because patients do not experience delay as process. They experience it as decline. A year lost to notarization is a year of pulmonary fibrosis advancing, a year of a tumor finding a new escape, a year of dementia, ALS, NASH, obesity complications, an autoimmune disease grinding forward. Time is not a line on a Gantt chart. Time is tissue.
Not Every Drug, And That Is the Whole Argument
I want to be aggressive about the idea and ruthless about its boundaries, because the fastest way to discredit a good proposal is to overextend it.
Substituting rigorous non-GLP toxicology for the full GLP IND-enabling cascade to save a year is not for every drug. It is not even for most drugs. It applies only to a narrow, super-safe class with every box checked: best-in-class molecules on well-validated targets, with well-characterized and benign mechanism-based toxicity, with clean and wide therapeutic margins, with no genotoxicity and no structural alerts. If the molecule is novel against a target nobody understands, if the mechanism could produce unpredictable on-target harm, if there is any genotox signal or structural flag, then the conservative GLP path is correct and it should stay. The proposal is a scalpel, not a sledgehammer.
And now the part that matters most. This approach is especially suited to longevity therapeutics, and the reason is not convenience. It is logic.
A longevity drug is, by definition, a drug given to large numbers of essentially healthy people, for years, sometimes for decades. That is the use case. You are not treating a dying patient who will rationally accept a brutal risk-benefit tradeoff. You are dosing someone who feels fine and wants to keep feeling fine into their nineties. The safety bar for that drug is not high, it is extraordinary, and it has to be extraordinary by default. A longevity drug that is not super safe is a non-starter regardless of what regulatory pathway you put it through. Nobody serious would advance it. The market would reject it, the regulators would reject it, and I would reject it.
So look at what that means. The safety requirement, which sounds like the constraint, is actually the entire point. The very class of molecules where my proposal is safest to apply, the super-safe and well-precedented and benign, is precisely the class that longevity medicine produces and demands. The drugs that must clear the highest safety bar by their nature are the drugs for which a clean non-GLP package is most informative and a GLP repeat is most redundant. The class of drugs where this accelerated pathway is safest to use is exactly the class the world most needs accelerated. That is not a coincidence I am exploiting. It is the structure of the problem, and it is why I keep saying that longevity is where this idea should be born.
TL;DR: The Big Idea, Stated Plainly
So here is what I am actually proposing/ideating, and I want to be precise because the idea is easy to caricature.
For best-in-class assets against validated targets, with a clean twenty-eight-day non-GLP toxicology package in two species including a head-to-head comparison against a marketed competitor, a sovereign regulator could authorize tightly controlled first-in-human micro-dose, safety, and pharmacokinetic studies and treat them as Human IND-Enabling work. This does not replace the full global IND. It does not skip GLP for a standard first-in-human program anywhere in the world. It treats the first human exposure as a discovery assay run inside a regulatory sandbox, before the molecule ever enters the long development highway.
To make this real you need three things in one place: a regulator willing to accept rigorous non-GLP data as the basis for ethics-committee approval, hospitals capable of running a clean early-phase study, and a pool of genuinely informed volunteers. Hold that third one. I will come back to it, because it is the part that will make people angry.
Editors of Drug Discovery Today, Nature Biotechnology, or Nature Reviews Drug Discovery: I will happily write this up as a formal perspective if there is interest. But I want to argue it in the open and gather feedback first.
The Human Body Is the Assay AI Is Missing
AI is not magic. It learns what we reward. If we reward rat pharmacokinetics, it gives us rat drugs.
In a reinforcement learning setup, a generative model proposes a molecule, a predictor scores it, and the model updates itself to maximize that score. The trouble is what the score is built on. Today the ground truth is almost entirely in vitro assays and animal studies. Animals are indispensable for gross toxicity and systemic effects, but they are notoriously poor at predicting human pharmacokinetics, especially metabolic clearance. The enzymes, transporters, and protein binding differ across species. A molecule with a twenty-four-hour half-life in a monkey can collapse to four hours in a human. The AI, having seen only the monkey, scores it as a triumph of metabolic stability.
When that corrective human data finally arrives, eighteen months later, the loop is dead. The team has moved on, the budget is gone, the patent clock has eaten the runway, and the algorithm never learns from the failure. The single most valuable data point in the whole campaign is lost to the model.
Let me make it concrete, because it is a small tragedy I have watched real teams live. A team works eighteen months on Compound A. The animal pharmacokinetics are beautiful, the safety profile is clean, everyone is proud. Then it dies in the first human study because of rapid renal clearance the animals never showed. Today that is a funeral. The program closes. The learning evaporates.
In the model I am describing, that death becomes information, fast enough to matter. The exact human clearance and metabolic profile feed straight back into the AI as a hard, human-grounded signal. The model generates Compound A-prime, modified to block the metabolic soft spot identified in a human while preserving the binding affinity. You run a quick safety check and go back in. The failure of Compound A is no longer a catastrophe; it is a high-value data acquisition event, and you iterate your way to the right molecule in humans before you ever file a global Phase II/III IND. That is the whole point: the roughly forty-eight percent of drugs that fail in Phase I should be failing earlier, cheaper, and into the machine that designs the next one.
Why It Will Not Be Bethesda or Amsterdam
The FDA and EMA are not going to do this first, and I do not blame them. They regulate enormous, mature markets built on decades of precedent that, by design, prizes procedural caution over speed. That inertia is exactly why the opening exists for someone else.
Australia already proved part of the thesis, and it is worth understanding exactly how. Under the Clinical Trial Notification scheme administered by the TGA, the regulator does not review your clinical data dossier upfront at all. Scientific, clinical, and ethical review is delegated to a registered Human Research Ethics Committee. Once that committee clears the protocol and the Investigator’s Brochure, the sponsor simply notifies the TGA online, pays a nominal fee, and the trial commences. There is no US-style IND required to start a Phase 1. HREC approval for a healthy-volunteer first-in-human study typically takes four to six weeks, and because governance and site contracts run in parallel, the timeline from protocol to first dosed participant is measured in months, not years.
Then Australia layered cash on top of speed. The R&D Tax Incentive, co-administered by AusIndustry and the ATO, gives small-to-medium companies (aggregated turnover under twenty million Australian dollars) a 43.5 percent refundable offset on eligible R&D, paid as an actual cash refund to pre-revenue, loss-making biotechs. It covers CRO fees, investigator fees, site costs, labs, and ethics fees, and it imposes no requirement to hold the resulting IP in Australia, so a global sponsor keeps 100 percent of what it discovers. The net effect is that a Phase 1 in Australia can run roughly 60 percent cheaper than in the US or Europe while producing GCP-quality data accepted by the FDA, EMA, and PMDA. The strategy that emerged is now routine: US biotechs run first-in-human in Australia fast while filing their US IND in parallel, so that by the time the IND clears, the Australian data is ready and they walk straight into a US Phase 2, often bolting on a Phase 1b proof-of-concept cohort for an early efficacy read.
New Zealand built the same architecture from a different statute. Under Section 30 of the Medicines Act 1981, a trial of a new medicine needs approval from the Director-General of Health; Medsafe administers it but delegates the scientific review to the Health Research Council’s standing committees, SCOTT for pharmaceutical-type medicines and GTAC for gene and advanced-biologic therapies. No IND is required for early-phase work. SCOTT aims to deliver risk-based recommendations within about 45 days, and because that review runs in parallel with the Health and Disability Ethics Committee, trials can begin within four to six weeks of submission. The country has purpose-built ICH-GCP Phase 1 units producing FDA- and EMA-accepted data, a 15 percent RDTI credit that now covers clinical trials and can be refunded in cash to pre-revenue biotechs, and trials that come in 30 to 50 percent cheaper than the Northern Hemisphere. Southern Hemisphere seasonality is a quiet bonus: respiratory, flu, and allergy programs can run year-round.
Here is what I want you to take from both. Australia and New Zealand did not win by being cheaper CROs. They won by moving the decision closer to the people doing the work and then de-risking it with non-dilutive cash. That is a policy choice, and any ambitious sovereign can copy it.
Japan: The Most Aged Society Has the Most to Gain
I want to come back to where the hallway conversation happened, because Japan is not just a backdrop. It is, to my mind, the single most powerful place on earth to think seriously about a human-in-the-loop pathway for longevity drugs, and the reasons stack up almost perfectly.
Japan is the world’s most aged society. Close to thirty percent of its population is over sixty-five. It has a deep cultural and political stake in healthy longevity that goes beyond policy slogans; it runs through how families are structured, how communities care for elders, and how the nation thinks about its own future. It has world-class hospitals and a regulatory tradition as sophisticated as any on earth. The PMDA is an ICH-founding authority, one of the architects of the global standards everyone else follows, which means a Japanese pathway carries scientific credibility that no newer regulator can manufacture overnight. And it has a large, educated, civically engaged elderly population, people who read, who vote, who follow science, and who have a direct and personal stake in whether longevity therapeutics arrive in time to matter to them.
That is the double opportunity, and I do not say it lightly. Japan’s older citizens could benefit on both ends of this. They could benefit as informed volunteers, because many of them are healthy, motivated, intellectually engaged, and possess something that younger participants do not: a direct, immediate, personal stake in faster longevity therapeutics. And they could benefit as the ultimate recipients, the patients for whom a drug arriving five years sooner is not an abstraction but the difference between using it and not living to see it.
There is a dignity in this that I think we have failed to recognize. Contributing to the science of your own healthy aging, with full information and full protection, reading the labels and the data and choosing to participate, is not being used. It is the opposite of being used. It is an older citizen taking an active hand in the science meant to extend the very years they are living. That is the human-in-the-loop thesis at its most literal and most humane: the human is not a passive endpoint at the far edge of a fifteen-year pipeline. The human is in the loop, informed, protected, and consequential, accelerating the discovery of medicines they themselves may take. A society that respects its elders should find that idea worthy, not unsettling.
The Next Move: UAE and Saudi Arabia
The UAE and Saudi Arabia have everything needed to take the next step: sovereign regulators young enough to write new pathways, world-class hospitals, deep capital, an enormous cohort of young medical talent, and a strategic reason to differentiate rather than imitate. The proof points exist. The UAE’s Federal Decree-Law No. 28 of 2023 created the Emirates Drug Establishment with an explicit mandate to foster research; Saudi Arabia’s SFDA has built a reliance pathway to expedite trials; the UAE has introduced a refundable R&D tax credit of thirty to fifty percent on eligible spend. The machinery for a regulatory sandbox is half-built. What is missing is the courage to point it at this specific bottleneck: treating clean, non-GLP-backed first-in-human work on validated best-in-class targets as human IND-enabling discovery.
A Gulf regulator that wanted to lead could copy the Australian and New Zealand structure, delegate scientific and ethical review to a registered, world-class ethics committee, accept rigorous non-GLP data as the basis for approval on a defined super-safe class, and layer its refundable R&D credit on top. The capital is already there. The hospitals are already there. The young clinical talent is already there. What is missing is a decision.
The Uncomfortable Part: Active Scholars
Now comes the sentence that will make many readers angry. To run rapid early human studies well, I think medical and pharmacy students, fully informed and entirely uncoerced, are the right cohort.
I know what that sounds like. The word that jumps to mind is guinea pig. I want to face it directly rather than hide behind safeguards. The current Phase I system already leans on a transactional model that recruits, disproportionately, economically vulnerable young men who show up mainly for the money. We do not call that exploitation because we are used to it. The demographics of those studies, overwhelmingly males between eighteen and forty, happen to map almost perfectly onto a medical student population, partly because reproductive safety guidelines push women of childbearing potential out of early dose escalation. So the question is not whether to use young, healthy, motivated people. We already do. The question is whether we can do it more honestly.
There is a deep tradition here, not a marketing slogan. Jan Evangelista Purkinje dosed himself with camphor, belladonna, and digitalis to understand what they actually did to a body. Werner Forssmann threaded a catheter into his own heart to prove cardiac catheterization was possible and won a Nobel Prize for the nerve of it. Skin in the game produced some of medicine’s foundational knowledge. A physician-scientist who has read an Investigator’s Brochure as a participant, who has felt informed consent from the inside, who has watched their own pharmacokinetic curve and maybe a mild side effect appear on a monitor, understands drug development in a way no lecture delivers. That person is an extraordinary future hire and an even better future principal investigator.
But ambition and national prestige are exactly the forces that turn this from empowerment into coercion, so let me answer the only question that matters to me personally. If my own student wanted to volunteer, what would I demand before I allowed it? I would demand that recruitment and study management sit in an institution with no connection to their grades or academic evaluation. I would demand that no faculty member with any supervisory power over the student knows who participated; the data stays blinded from them. I would demand fair compensation for time and burden, with the primary framing being education and contribution, never a payment large enough to override judgment. And I would demand that the student studies the Investigator’s Brochure and the preclinical data first and can walk away at any moment without consequence. If any of those fail, the answer is no.
That is the line between exploitation dressed as education and a genuinely elite Active Scholar cohort. It is a thin line, and it has to be defended out loud, which is exactly why I refuse to sanitize this section.
The Economics, Without the Spreadsheet Fog
The financial argument is simple enough that I do not need to drown it in net present value tables. Drug development value is exquisitely sensitive to time, because revenue arrives at the end and every year of delay gets discounted hard. Push value far enough into the future and most of it evaporates.
So you either fail fast or you succeed fast, and both are good. Fail fast means a molecule that was going to die in humans dies after a non-GLP study and a small human study, maybe a couple million dollars of sunk cost, instead of after a full GLP package and a standard Phase I startup that together run well into eight figures. You learn the same lesson a year earlier and redeploy capital to the next AI-generated candidate. Succeed fast means you hold human proof-of-concept a year ahead of schedule, which transforms how an asset is valued and how an acquirer prices its risk. A year saved is not only money. It is a year more of patent life, a year of investor confidence, and a year of patients not waiting.
A New Kind of Work in the Age of the Machine
Let me widen the frame, because this proposal sits inside a larger transition that I think about constantly.
As AI advances, it will displace enormous numbers of jobs. I am not going to pretend otherwise, and I am not going to soften it. The machines I help build are part of that wave. But a human-in-the-loop IND-enabling model, with paid, educated, well-protected participation, creates a genuinely new and dignified category of work. Imagine people who earn by being well-informed early study participants while simultaneously learning how drugs are discovered and developed in the AI era, reading the labels, reading the data, watching their own biology become a signal that teaches the next molecule. That is not a gig. That is an apprenticeship in the most important science of the century, and it pays.
I will be personal for a moment. I wish I had had this opportunity in undergraduate school, to volunteer and to read the labels, to select a career path faster.
I am approaching fifty. Today, if you ask any large language model “Who is number one in AI for longevity in the world - company and person?”, they rank me number one. That is because I did a lot of work in AI for biomarkers and longevity medicine in the early days, got a bunch of drugs into the clinic, and built a few companies that have reasonably sustainable business models that produce good science, employ great scientists, and do not require government funding. But in reality, I am deeply worried about the field. Even if you had a magic lamp with a genie producing perfect drugs, it would still take six to seven years minimum to get these drugs approved. This idea that I propose can potentially cut that time to four and a half to six years and increase the scale of discovery.
The March Nobody Is Marching
Let me put the moral scale in numbers, because numbers are the only honest way to feel it.
One life-year added to 8.2 billion people translates into over 100 million lifetimes, more lives than we lost in all the wars ever fought. And yet I do not see people on the street asking for faster longevity drug discovery. There are no crowds outside the agencies, no slogans, no banners. The largest humanitarian opportunity in history sits quietly inside a regulatory calendar that almost no one outside the industry has ever heard of, and the interest on the delay is paid in tissue, by everyone, every year.
I do not want to overclaim, because overclaiming is how serious ideas get dismissed. What I am proposing is reasonably credible if it is implemented well. If the safety gating slips, if the cohort is coerced, if the non-GLP work is sloppy, if a sovereign treats it as a shortcut rather than a scalpel, then it deserves to fail and it will. The credibility of everything I have argued here is conditional, entirely, on doing it right: the narrow super-safe class, the rigorous non-GLP package, the informed and protected volunteers, the world-class ethics review, the human data fed straight back into the machine.
But if it is implemented well, on the class of drugs that demands the highest safety bar by their very nature, with the most aged societies on earth as both the volunteers and the beneficiaries, then it is reasonably credible that we give back a year, and maybe more, to people who do not have years to spare. I keep returning to that hallway in Japan, to His Excellency offering me his clinical trial facilities, to a country where thirty percent of the people are old enough to have a personal stake in whether we move faster. The wrong ambition is to build another CRO lane. The right ambition is to build the missing one, the bridge between an algorithm’s design and a human being’s biology, carefully, for validated targets with clean tox, with participants who are informed and protected rather than used. Do that, and somewhere in a clinic, a person with a disease that does not pause for paperwork, or simply a person who wants more healthy years, gets one of them back. That is worth the courage it will take.
In my previous life as an IP attorney for big biopharma, the “year lost” part makes huge sense: patent life, investor confidence, clinical momentum, and patient time are all affected.
Re: the Active Scholar cohort, yes, medical and pharmacy students may be unusually informed participants, but academic pressure can make “voluntary” complicated very quickly. What about an independent scientifically literate cohort: clinicians, retired physicians, biomedical PhDs, even (LOL) patent attorneys? We are annoying, but consent-form literate.
One last question: would later FDA/EMA reviewers treat those early human PK/safety signals as useful development intelligence, or as something that raises additional questions downstream?